Richard E Kaye Annotation 74 alr Fed 2d Art 1 2018
Method Article
Revised
StateHub-StatePaintR: rapid and reproducible chromatin country evaluation for custom genome annotation
[version 2; peer review: 3 approved with reservations]
Writer details Author details
Open PEER REVIEW
REVIEWER Status
Introduction
Chromatin segmentations are increasingly important for a broad expanse of inquiry that includes regulatory genomics, genetic epidemiology, precision wellness, and molecular genetics. In that location is a need for consequent, unbiased resolution of chromatin states to interpret the epigenome and predict role across different tissues and cell types.
Complex, overlapping patterns of post-translational modifications (PTM) to histone subunits1,two, signify differing states of chromatin activity. These modifications consist of mono-, di-, or tri-methylation and acetylation of histone 3 lysines 4, 9, 27, and 363. Straight assays for histone PTMs with next-generation sequencing (NGS) using chromatin immunoprecipitation (ChIP-seq) event in a set of genomic intervals with evidence for enrichment over background (input chromatin), using signal intensity.
In addition to ChIP-seq of histone PTMs, there are also NGS methods for histone displacement, including DNase I hypersensitivity4 (DNase-seq or DHS), Formaldehyde Assisted Isolation of Regulatory Elements5 (FAIRE-seq), Assay for Transposase Accessible Chromatinsix (ATAC-seq) and Nucleosome Occupancy and Methylome sequencing (NOMe-seq)7. Histone displacement, nucleosome positioning and Dna methylation are also detected in genomewide assays (east.1000. whole genome bisulfite sequencing8). Histone displacement is associated with transcription factor bounden and transcriptional activityix. In improver, direct binding of transcription factors is measured in ChIP-seq experiments with an antibody directed against a transcription factor or an epitope-tagged version.
All these data are compatible with data represented equally genomic intervals (in BED format), including CpG islands, annotated transcription commencement sites, repeat elements, 3′ UTRs. The input and final (output) processed data format are both represented every bit browser extensible information (.bed), a flexible standard for different peak calling methods (east.thousand. "narrowPeak" and "broadPeak" are types of .bed files).
Several car-learning approaches integrate NGS experiments into annotation tracksx. The goal is to discover epigenomic states and aid in agreement "non-coding" genomic elements in an unbiased and biologically meaningful manner. Newly discovered states are an constructing of true functional categories of chromatin biological science. The most popular and widely used of these machine learning methods is ChromHMM11. Other machine-learning approaches include spectral-based learning12, inference based on read counts13, dynamic bayesian networksfourteen, probabilistic approaches15, supervised enhancer detectionxvi, and other hidden Markov methods17–19.
The interpretability and general usefulness of the state predictions produced by these algorithms varies. A multitude of states often must be consolidated into simpler, biologically meaningful categories. Hoffman et al., recognized this trouble when they proposed a combined meta-assay of ChromHMM and Segway annotations20. However, a software framework for adept or rule-based segmentations is still lacking. Comparisons beyond heterogeneous data sets, involving different learned models, or slightly different sets of epigenetic marks, must be performed advisedly, tracking how annotations are created and which tin exist considered uniform. In addition, it is necessary to update information virtually what annotations are appropriate equally new evidence about the combinatorial patterns of the epigenome come up to light. Such methodology is needed for integrating different experimental information (including not-NGS information) in a reproducible fashion, reflecting both the novel insights gained from the machine learning methods and our current understanding of genome biology.
Hither we introduce StateHub and StatePaintR for generating and documenting chromatin state and other genome sectionalisation models in a transparent and reproducible fashion. StateHub is a customs resource for storing annotation models, state definitions and associated data in a shareable, referenceable form. The StatePaintR package implements these models and land definitions to produce notation tracks based on histone and other epigenomics marks, sequence features, and gene annotations. We prove that StatePaintR can exist used to rapidly comment big collections of public data for summarizing epigenomics data or annotation of variants. We bear witness how annotations gracefully degrade, in that cell types or tissues with missing data types are annotated appropriately based upon available information. We show some use cases and describe how StatePaintR uses Fleck-seq data elevation statistics to rank the state prediction for each segment. The priority of the method is to provide a framework to express existing statements about the relationships of genomic annotations and how they combine to reveal underlying chromatin states thereby bypassing denovo learning and annotating of states within each sample and annotating solely based upon simple rules and bachelor data.
Methods
Implementation
StatePaintR is implemented as a software package in the R language freely bachelor from the Bioconductor repository: www.bioconductor.org/packages/release/bioc/html/StatePaintR.html. The package contains functions for generating notation tracks from called peaks specified as intervals co-ordinate to the rules specified in a decision matrix and an abstraction layer describing the relationships betwixt specific assays and functional categories. An abstraction layer may define a single functional category for a collection of assays that represent like biology, e.k. assays for H3K27ac and H3K9ac may both represent an "Agile" functional category. These data are supplied to StatePaintR in the class of BED files, or one of their extensions (e.thousand. narrowPeaks, gappedPeaks), leaving it to the user to either telephone call areas of enrichment/peaks in the manner they call up best, or acquire pre-called peaks from a trusted source. The decision matrix encodes the relationship between these functional categories and specific chromatin states, where the values of whatever particular cell of this matrix must take whatsoever of 4 dissimilar values (Table ane) indicating the nature of the relationship. Together the abstraction layer and the decision matrix describe a StatePaintR model.
Table 1. StatePaintR matrix values.
StatePaintR assigns annotations according to custom rules specified in a matrix. The rules are represented as an integer code that takes whatever of iv values [0–3]. The pregnant of each value is summarized in the table.
| required or state? | consistent with state? | binary value | decimal value |
|---|---|---|---|
| No | No | 00two | 0 |
| No | Yes | 012 | 1 |
| Yep | No | 102 | 2 |
| Yeah | Yes | eleven2 | 3 |
Each jail cell of the conclusion matrix relates functional category to chromatin land in a 2-bit lawmaking representing the answers to 2 TRUE/Imitation questions (see Table one). Is the functional category required in guild to telephone call the land? And, is overlap consistent with the state? For the purposes of explanation, examples beneath use the nomenclature of our "focused poised promoter model", but a user may create their own model or modify the decision matrix or abstraction layer of an existing model. The cell of the decision matrix defining the relationship between the country "Poised Promoter Region" (PPR) and the functional category representing narrow meridian calls of H3K27me3, "PolycombNarrow" is 3, representing the binary value 11two. This encoding indicates that in lodge to call the PPR state on an interval, data representing the "PolycombNarrow" functional category is required to be nowadays, and second, the interval in question must also overlap with a peak described by that functional category. A score of two representing the binary value ten2, as in the cell describing the relationship between PPR and the functional category "Agile", indicates that in guild for the interval to be annotated as PPR, data relating to "Active" must be nowadays in the data prepare, simply must non overlap the queried interval. A score of 0 representing the binary value 002, as in the prison cell for the functional category "Core" (which incorporates DHS, ATAC-Seq, and FAIRE meridian calls) and PPR, indicates that it is not necessary for data represented by "Cadre" to be present, however if the "Core" data is present and overlapping the queried interval, the PPR country cannot be called. The category "Translation marks" does not affect PPR in this model, even if it overlaps. Marks that are substantially irrelevant to PPR such equally this one are assigned 1 representing binary 012.
Thus established, each row (as "state") in the determination matrix is a unique combination of values describing the relationship of the functional categories to the state, where the rows are organized past the software in club of state complexity. StatePaintR starting time generates a GRanges list (an R object containing a list of chromosomes and interval coordinates with capricious metadata columns attached) of all uniquely mapping segment boundaries from the start and end coordinates of every peak in all files. StatePaintR then evaluates the presence or absence of each functional category and eliminates erroneous states. Side by side the program assesses overlaps of each segment to determine whether the conditions specified in each cell of the decision matrix are compatible with that segment, producing a boolean value. Rows with perfect matches in all cells are candidate state calls. Since StatePaintR evaluates in order of increasing state complexity, lower complication states can be overwritten if higher complexity states match. This is very useful for building degeneracy in a model. An example of this in Figure 1 is illustrated by us, ER and EAR. If active marks (e.g. H3K27Ac) are not available for a given cell type, StatePaintR volition annotate H3K4me1 marks as ER under our default model. In a dissimilar jail cell type for which H3K27Ac information are available, StatePaintR will know to distinguish between H3K4me1 enriched regions as either active or poised based on overlap of this second marker. Thus, a model tin specify different state calls equally appropriate based on the availability of data for each cell type. StatePaintR includes a peak score for each state drawn from all experiment categories (columns) that accept a matrix value of 3, i.e. because they are required for and consistent with that state. The acme scores are rank normalized on a scale of i to i,000, with one beingness the minimum peak size and 1000 being the maximum. If multiple categories are required, StatePaintR selects the median peak score for the annotation. This behavior can exist overridden (see documentation for details).
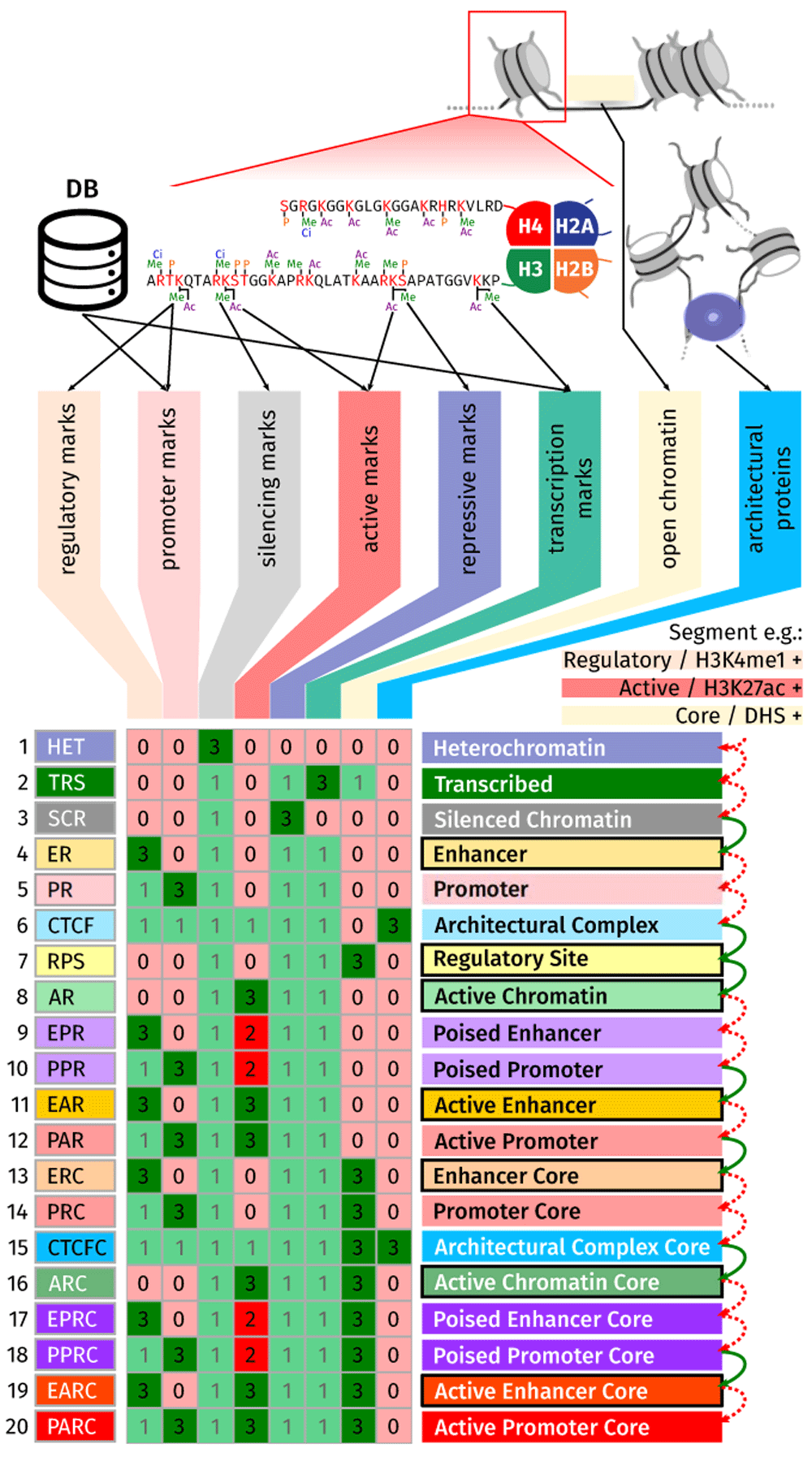
Effigy 1. Mapping datasets to functional significance annotations.
Experimental data and external database annotations are combined into abstraction layers (columns), integrated to produce chromatin states (rows) from the conclusion matrix. StatePaintR produces state assignments by iteratively comparing the marks that are nowadays in each segment with each row of information in the tabular array. The values of color-coded squares signify relationship betwixt data and land: 0 (low-cal red) the characteristic/data type negates the state but is not required to be present, 1 (lite dark-green) feature is consequent with the state but not required, 2 (ruby) if the characteristic is required to be available and negates the state, and 3 (green) it is both required and consistent with the country. Complication of states increases from acme to lesser. For the example, red dotted arrows, proceeding downward, point to non-matching rows, and green arrows point to matching rows. The country call corresponds to the last matched row. In this example with the presence of H3K4me1 ("Regulatory"), H3K27ac ("Agile") and DNase1 hypersensitivity ("Core"), the starting time state consistent with the presence of these functional categories is "Enhancer", followed past the increasingly more complex "Regulatory Site", "Active Chromatin", "Active Enhancer", "Enhancer Core", "Active Chromatin Core", and finally "Agile Enhancer Cadre".
Finally, once all segments are annotated, and scored, StatePaintR is able to export these annotations as BED files that may be viewed in whatever genome browser. The package includes an R-markdown vignette. The current release version of this vignette is always available from the Bioconductor website.
StateHub is implemented equally an interactive website (world wide web.statehub.org). StateHub contains a database implemented in MongoDB and a search engine written with Google Web Toolkit (GWT), which updates dynamically with user input. This database includes all models, model metadata and pre-computed StatePaintR browser tracks. Models are composite JSON objects that include an unique identifier, name, revision number, a searchable text description, and a model matrix (as defined in Table 1). The website also includes links to this manuscript, R-markdown containing code for figures, the latest version of the vignette, links to twitter feed and additional instructional materials.
StateHub models
The main text makes reference to two models in StateHub (statehub.org). The unique identifiers of these models are equally follows: "Default" (model ID: 581ff9f246e0fb06b4b6b178) and "Focused Poised promoter" (model ID: 5813b67f46e0fb06b493ceb0). In each of the two models presented and discussed in this paper nosotros chose a naming convention for our states reflecting biological function.
Note of public datasets
Preprocessed peak calls were obtained from the IHEC and ENCODE websites (see Table two) for hg19, and where possible hg38. Where possible nosotros used IDR (Irreproducible Discovery Rate) candy narrowPeak calls for DHS and broadPeaks for broad marks (H3K27Ac, H3K4me1, H3K27me3, H3K36me3) unless otherwise specified in the model. A complete manifest with filenames, plus all annotation tracks are bachelor on the StateHub website.
Table 2. Annotation of public datasets.
Data from the indicated public consortia were downloaded and processed in StatePaintR. The resulting annotation files and browser sessions are available from the StateHub spider web page under each model page.
| hg19 | hg38 | mm10 | |
|---|---|---|---|
| Blueprint (IHEC) | 630 | 548 | 0 |
| CEEHRC (IHEC) | 158 | 0 | 2 |
| DEEP (IHEC) | 22 | 0 | half dozen |
| ENCODE | 84 | 109 | 98 |
| Roadmap | 127 | 0 | 0 |
Enrichment calculations
Parkinson'south GWAS variants. To illustrate the use of StatePaintR chromatin state segmentations in GWAS functional annotations, nosotros revisited an earlier study of Parkinson'due south disease in which we tested for tissue-specific enrichment of genetic associations. Parkinson'southward GWAS variants were obtained from a previously published large scale meta-analysis21. Nosotros used a beta-binomial conjugate distribution to estimate the apparent range of differences in overlaps between observed (GWAS hits) vs. random variants. To summate enrichment nosotros selected all variants inside 1 MB of the index SNP in each region with a minor allele frequency (MAF) > 0.01, defining foreground as SNPs in linkage disequilibrium with the index SNP at a cutoff of r 2 > 0.8 and background equally all SNPs inclusive (MAF > 0.01). Enrichment in genomic annotations. Analyses and graphics were produced using the SegTools package22.
Analysis of methylation data
To select methylation variants, we analyzed the Infinium HM450 data of 114 ovarian tumor samples23 and 216 command normal Fallopian tube samples24. We define differentially methylated regions as those having a difference in beta values of 0.three (cancer vs. normal) and significance in Mann-Whitney U-test (FDR-corrected p-value < 0.01). We then performed enrichment calculations using overlaps between probes that were hypermethylated in cancer vs. normal and the state calls from two models described above and in the text. The enrichment calculations were washed with fisher'south exact test using the consummate HM450 probeset as groundwork.
Operation
All code used to generate figures, tables, and this manuscript is included as an R-markdown certificate (Supplementary File ane)25. A copy of this document may also exist obtained from the StateHub website. In addition, a workflow vignette is available from the bioconductor package and mirrored on the github repository at github.com/Simon-Coetzee/StatePaintR.
Results
A framework for rule-based annotation
In order to assign chromatin states, it is necessary to account for the complex interplay of input from genomic annotations and cell-type-specific experimental data sources that ascertain and demarcate functional regions of the genomeone. Computationally they have to exist put in the right order to avoid erroneous overwriting of information-rich categories with information-poor ones.
Nosotros initially wrote a model every bit a decision tree, encompassing a fix of bones rules for annotation, but this approach was limited in that any pocket-sized change to the model necessitated a near consummate re-write of our software. Secondary to this, we wanted a solution that would enable u.s. to specify any modify in the model and accept it produced the same mode as all previous models while minimizing software updates. And thirdly, we felt that whatever such model should be reproducible, documented, citable and extensible to whatever combination of experiments. Moreover from a bioinformatics perspective, nosotros felt that any two colleagues working separately should be able to produce precisely the same annotations from the aforementioned datasets and models. To satisfy these unlike requirements nosotros separated the model specification from the note tool. We implemented model-specification as a conclusion matrix, which has the advantage of separating model specification from software, enabling complete explicit control of the annotation software without figurer programming expertise.
We created a searchable website, StateHub, to host a permanent repository of models, document model objects and brand them bachelor every bit a resource to the community. The StatePaintR package retrieves models from StateHub and performs annotations on local data. Thus, StateHub- StatePaintR is a framework to document models and utilise them to annotate genomic data. The models in StateHub consist of an brainchild layer, defining the relationships between data sources and functional categories. These categories are integrated to produce annotations (left hand cavalcade, "Chromatin States") via a decision matrix (Figure i). Within the model each state has associated descriptions of capricious length which may contain key words or other relevant details (bottom right).
Annotation scoring
StatePaintR enables rank scoring of all states, allowing prioritization for non-coding variant annotation. No other existing tool does both chromatin state notation and rank evaluation simultaneously. Thus, while automobile learning chromatin sectionalization methods are focused on characterization assignment alone, our paradigm preserves critical quality information from the underlying Scrap-seq data to arrive at overall rank scores. We used these rank scores to generate precision recall statistics for predicting experimentally validated enhancer regions from the VISTA database26. Our method outperformed most other methods aimed at predicting enhancers (Table 3). Different other methods, our tool did not rely on training data and non only was able to predict and score enhancer states, just whatsoever other arbitrary states that tin be described using the StateHub definition linguistic communication. No other existing tools provide this functionality with chromatin segmentation.
Tabular array 3. Relative performance of StatePaintR enhancer ranking vs. VISTA enhancers27.
Columns 2–6 reflect the expanse under the precision-recall gain (auprg) curve. Highest scoring algorithm noted with *.
| source | neural tube | mid-encephalon | hind-encephalon | limb | heart | average auprg | average rank |
|---|---|---|---|---|---|---|---|
| REPTILE | 0.86* | 0.87* | 0.76 | 0.89* | 0.92 | 0.86 | 2.0 |
| StatePaintR† | 0.84 | 0.84 | 0.79 | 0.85 | 0.88 | 0.84 | 3.0 |
| RFECS | 0.79 | 0.85 | 0.78 | 0.85 | 0.92 | 0.84 | iii.0 |
| ENCODE | 0.82 | 0.82 | 0.80* | 0.85 | 0.88 | 0.83 | three.iv |
| DELTA | 0.81 | 0.84 | 0.76 | 0.84 | 0.93* | 0.84 | 3.6 |
| CSIANN | 0.72 | 0.68 | 0.62 | 0.69 | 0.84 | 0.71 | 6.2 |
| EnhancerFinder | NA | 0.59 | 0.63 | 0.67 | 0.82 | 0.68 | six.8 |
Use cases
Segmentation of public datasets
We generated annotations of 119 ENCODE jail cell lines26, 128 Roadmap tissues28, 26 cell lines and tissues from CEEHRC (peak calls obtained from the IHEC website), and 23 blood cell types from Blueprint (download at statehub.org). On a desktop PC it takes approximately 12–15 seconds to produce an note from a typical cell line, depending on the number of datasets and intervals (see Figure S1). StatePaintR produces genome browser compatible BED files with colour-coded state annotations (specified in StateHub model). Effigy ii shows a representative region effectually the POLR2A cistron from a subset of 77 loftier-quality (minimum fifteen one thousand thousand reads) tissue samples and cell lines with H3K27Ac data from Roadmap. A complete manifest for processing these data is included in additional files one.
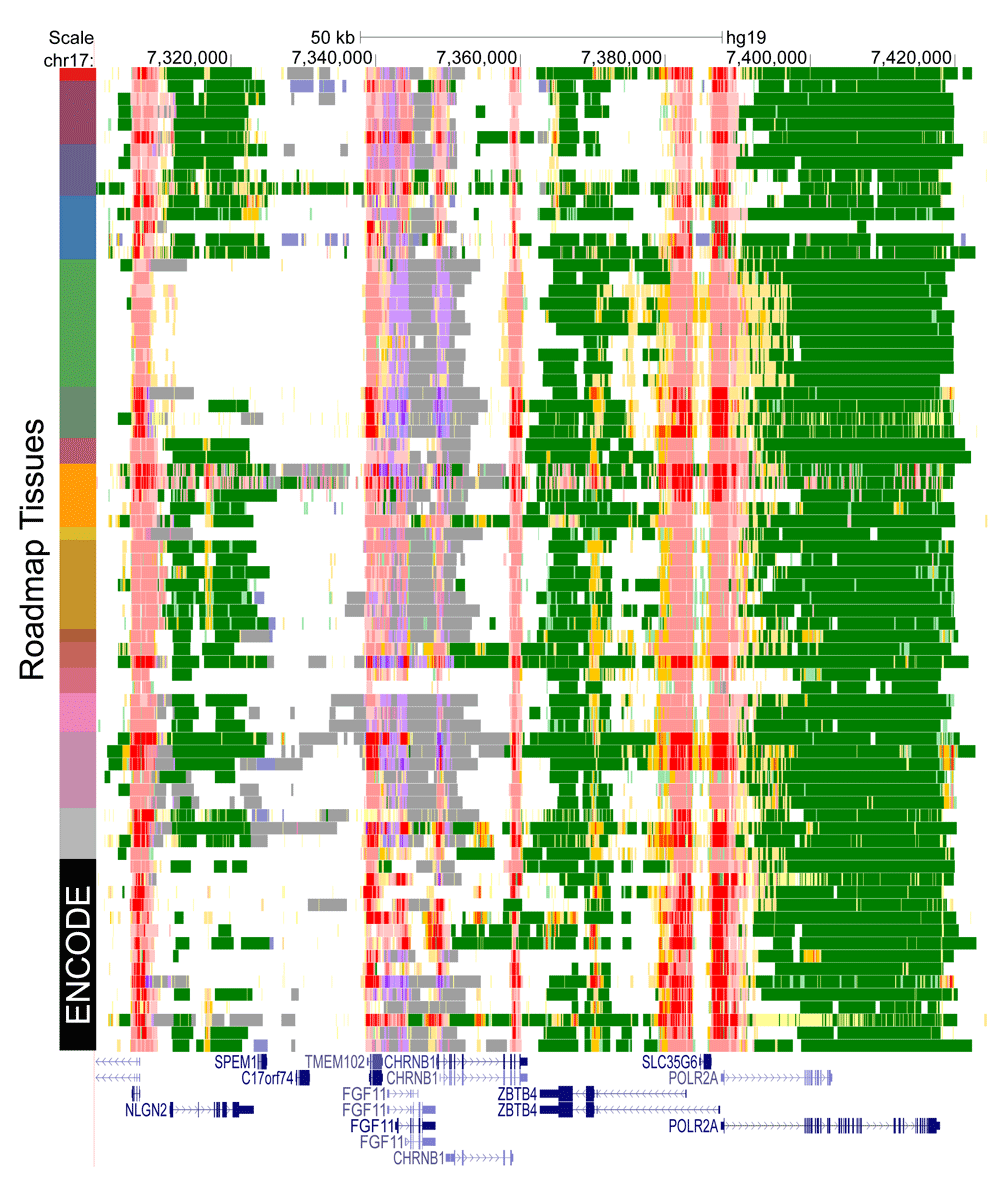
Figure 2. Notation of public epigenomics data sets.
Annotations of 77 prison cell types from the Roadmap Epigenomics consortium, including some Roadmap-processed ENCODE data, selected for their high quality with default model. Roadmap tissues are clustered and colour coded at left according to the aforementioned color scheme used in Roadmap publications28.
Annotation of genome-wide association studies
A common employ of genome notation is to assign putative role to genetic loci identified by genome-wide association studies (GWAS), peculiarly for non-coding regions. We previously used a custom annotation of Roadmap tissues based on the arroyo described in this manuscript to identify locus-specific tissue enrichment in variants associated with Parkinson's disease29. In that study, nosotros displayed locus-past-tissue enrichment as a heat-map. Here we present a similar analysis using our new StateHub model as the basis for an alternative visualization. Since nosotros showed that Parkinson's disease variants are primarily associated with enhancers and promoters29, nosotros plotted the 95% range of credible values for enrichment in enhancers and promoters vs background SNPs (matched for GC content & minor allele frequency). Each locus (row) is plotted against a selection of tissues in Roadmap (Figure 3).
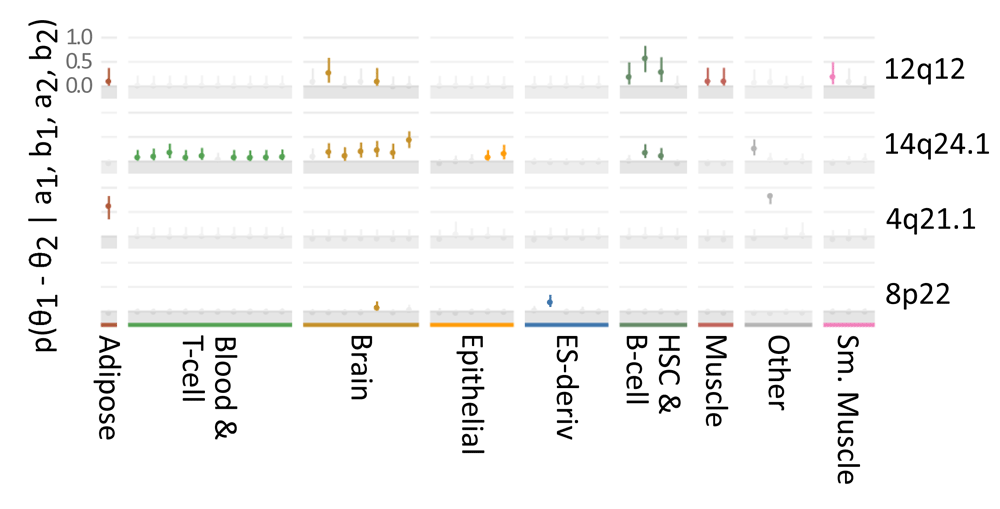
Figure 3. Locus- and tissue-specific enrichment of Parkinson's GWAS variants.
Confined: 95% credible range for enrichment of Parkinson's GWAS variants and LD proxies with R2 ≥ 0.8 in the spousal relationship of active enhancers and promoters vs SNPs in the region with similar pocket-size allele frequency and R2 < 0.8, for each of 4 independent genetic loci. θ 1, θ ii relative enrichment in foreground and background sets, respectively. a i, b one number of foreground SNPs overlapping biofeatures or not-overlapping, respectively. a 2, b 2 number of groundwork SNPs overlapping biofeatures or not-overlapping, respectively. a and b are shape parameters of a beta distributed prior. Significant enrichment profiles for roadmap tissues are displayed in colour (REMC lineage-specific colors); non-significant are grayness.
Evaluation of 2 models with respect to cancer methylation
Our "default" model proposes a form of enhancers and promoters in a poised state, an "Enhancer Poised Region" (EPR) and a "Promoter Poised Region" (PPR). These features have H3K4me1 or H3K4me3 and lack H3K27Ac. This model also classifies H3K27me3 as silenced/polycomb repressed (SCR). To investigate functional enrichment of methylation variants, nosotros looked at how differentially methylated regions (DMR) in ovarian cancer tumors partition between chromatin states every bit divers in this model (Figure ane).
From previous piece of work, CpG islands containing temporarily silenced (poised) genes by polycomb repressive circuitous in normal tissues may acquire DNA methylation during cancer formation resulting in permanent silencingxxx,24. While the segments called EPR and PPR were associated with hypermethylated probes in ovarian cancer across tissues, the magnitude of enrichment was not groovy (see Figure 4, "Model 1"), and it remained possible that our state definitions were likewise broad.
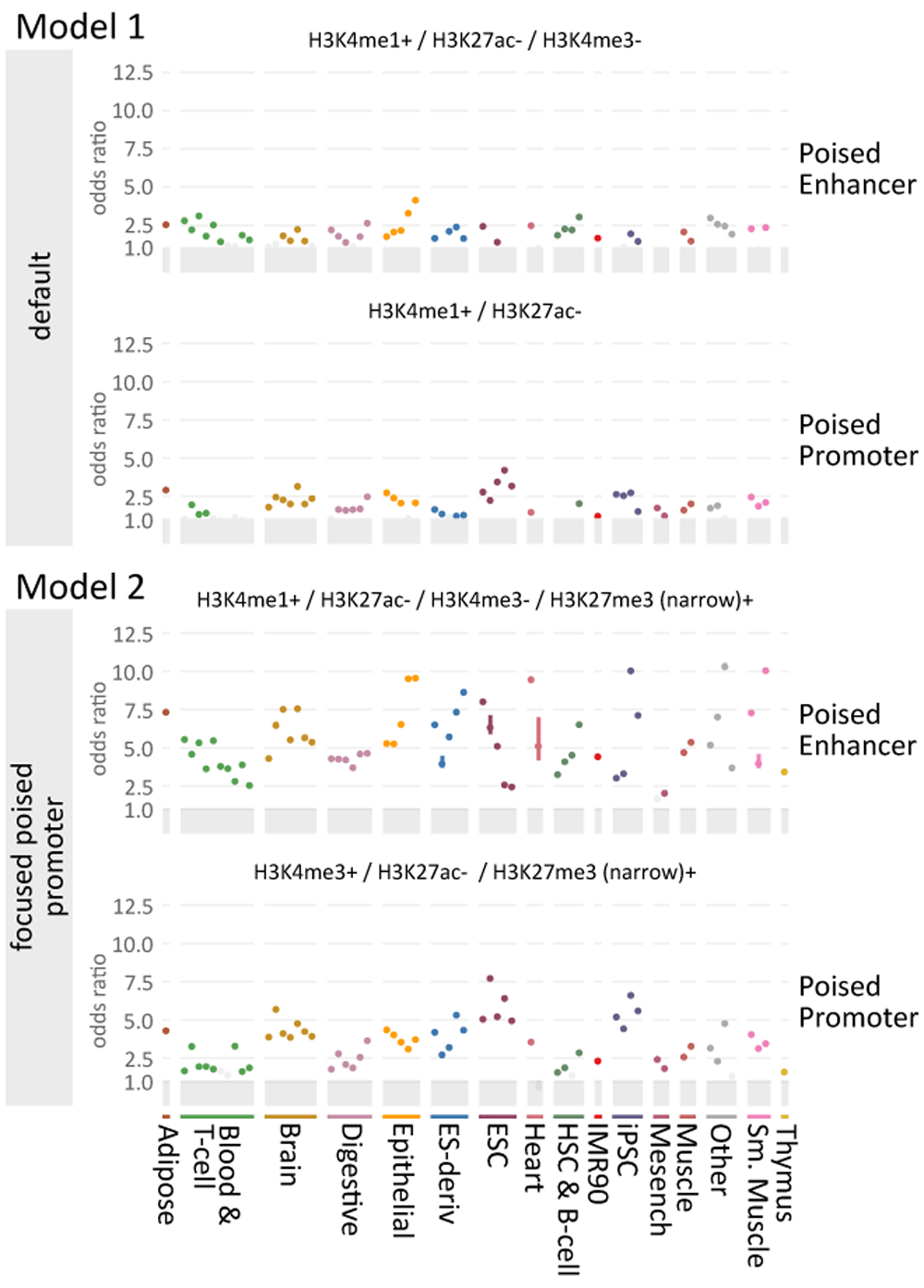
Figure iv. Case of model comparisons.
Enrichment equally in Effigy 3 using either of two different state models (model 1 and model ii) from StateHub, "Default" and "Focused Poised Promoter", which differ in the treatment of poised promoters. The clan of hypermethylated regions in ovarian cancer with poised enhancer ("Enhancer Poised Regions" – EPR) and promoters ("Promoter Poised Regions" – PPR) across roadmap tissues are indicated by odds-ratio in the Y-axis. Y-axis range is the aforementioned for both plots. Both models distinguish hypermethylated probes in the poised country but model two is more than selective than model 1. In this model (two) enhancers with H3K4me1 and promoters with H3K4me3 overlapping narrow regions of H3K27me3 are poised (EPR and PPR), only those without H3K27me3 are called weak (EWR and PWR). Model 1, past contrast, assigns promoters lacking active marks to the poised state.
I hypothesis is that poised promoters are distinguishable by the presence or absence of focused H3K27me3, in particular the narrowPeak calls (as opposed to broad, low-level enrichment from broadPeak files used in model 1). To address this hypothesis, we repeated the analysis in Figure 4 for an alternative model (model two; "focused poised promoter") in which H3K27me3 is called as both broadPeak and narrowPeaks. We utilise the H3K27me3 broadPeak file as in the previous model to identify repressed regions, and H3K27me3 narrowPeaks to identify poised states (EPR and PPR). Enhancers lacking H3K27Ac and H3K27me3 were classified equally weak enhancers and promoters ("Enhancer Weak Regions", EWR and "Promoter Weak Regions" PWR, non shown in Figure 4). Regulatory elements with these backdrop have also been called "primed"31.
We found greater enrichment when we defined poised states in this mode (compare model two (focused poised promoter) with model 1 (default) in Figure 4). The hypermethylated ovarian cancer CpGs were more than enriched in EPR, PPR, and SCR states equally divers in the focused poised promoter model relative to the default model, and hypomethylated probes were enriched only in HET and SCR states (not shown). The odds ratio of enrichment for hypermethylated CpGs in EPR and PPR from the default model fell in a range between 0 and 5. However, the enrichment of the hypermethylated probes in our focused poised promoter model was > five in PPR and > 10 in EPR (Effigy 4, model 2). Thus, ovarian hypermethylated probes are enriched across Roadmap tissues in H3K27me3+ enhancers and promoters, and nosotros ended that H3K27me3 narrowPeaks are an important distinguishing characteristic for this class.
Enrichment of functional annotation
Adjacent, we characterized the distribution of states in our focused poised promoter model relative to Gencode v37 gene annotations and also to enhancers from Ensembl32. Figure 5 shows the relative enrichment of Human mammary epithelial jail cell (HMEC) chromatin states in each of these features. We found enrichment in Ensembl enhancers for three states: Active enhancer (EAR), Active regions (AR) and Weak enhancer (EWR). The definition of "agile enhancer" in the Ensembl build is cumulative across prison cell types32 and therefore includes many cell-type specific enhancers that would be predicted to be weak (having exclusively H3K4me1) in a item cell line such equally HMEC. These three states were not enriched in any other category of genomic annotations. As well, we found enrichment of the inactive enhancers in Transcribed (TRS) and Silenced/Polycomb (SCR). TRS was near enriched in gene trunk annotations, peculiarly internal exons and introns. SCR and Heterochromatin (HET) were depleted across all categories. Lastly, the v′, get-go exon and first intron regions were enriched in active and weak promoters, consistent with the role of these regions in transcription initiation.
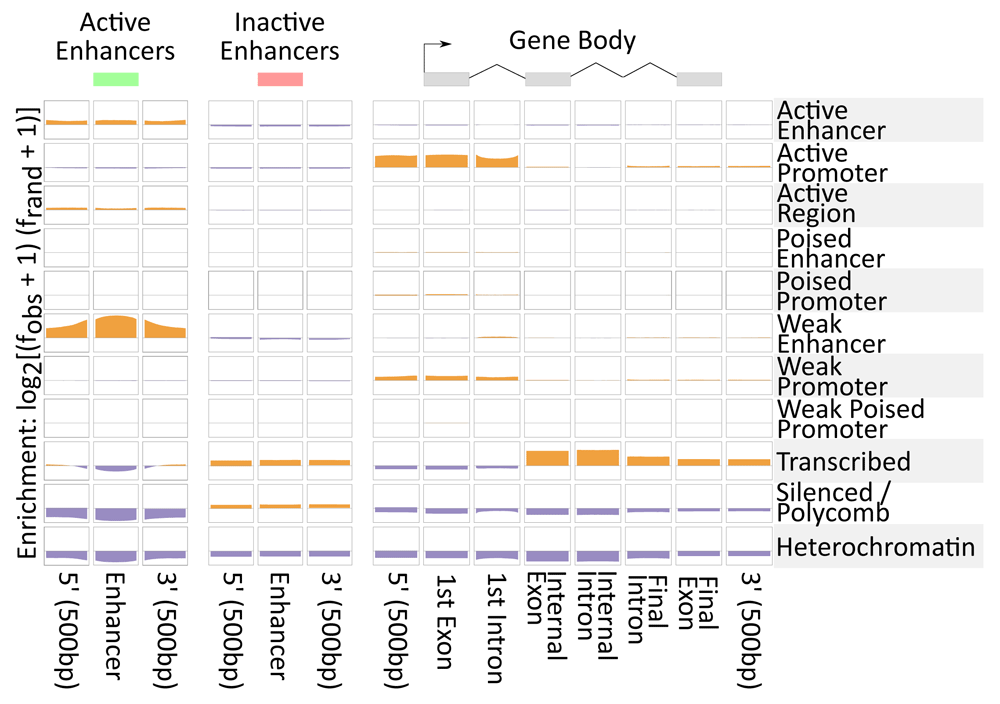
Figure 5. Enrichment in genomic annotations.
Relative enrichment of called states genomewide from HMEC in annotations from Ensembl and Gencode. Genegraph (meridian) visualization of the regions indicated for each column. Enrichment is log ii observed over random. Positive enrichment is indicated with mustard color (scale from 0 to 0.66) vs. relative depletion in purple (scale from 0 to -0.37).
Enhancer predictions
To utilise ChIP-seq data for quantitative analysis, we ranked within each state past peak score from Macs2 output (generic peak height). We programmed StatePaintR to rank each state by normalizing on a calibration of 1–thousand, 1000 being the highest rank. StatePaintR ranks the required dataset(southward) for each state (i.eastward. assigned "iii" in the decision matrix). To evaluate the ranking part, we measured area nether the precision-recall-proceeds curve (AUPRG) using the set of experimentally validated human and mouse noncoding fragments with gene enhancer activity as assessed in transgenic mice (VISTA enhancer browser and 27). We randomly sampled 100 enhancers from seven VISTA tissues to evaluate dissimilar aspects of our models (training), and and so used the residual of the data to exam our enhancer predictions against previously published predictions using the same information sets.
Some states, including the ones that are germane for enhancer prediction, reference more one required (matrix value 3) dataset, and therefore it was necessary to optimize the all-time method for ranking based on > 1 ChIP-seq experiment. We computed the average, median and ceiling functions of ranks across multiple ChIP-seq tracks. The three methods were comparable, but median and average produced the best results (Figure S2). There are three required marks for agile enhancers in our model, only if one of them is not informative for active enhancer prediction, using the ceiling "max" method would produce false positives when this mark has the highest summit rank. Therefore, we interrogated which marks are informative using a leave-one-out arroyo. Nosotros found that leaving out H3K4me1 significantly improved our predictions, whereas leaving out the other marks did not (Figure S3).
Next we assessed AUPRG of different land calls vs. VISTA enhancers and found that predictive ability descends in gild AR + EAR > EAR > AR > RPS > EPRC > etc (Figure S4). When nosotros tried combinations of states the highest precision recall proceeds was observed for EAR, EARC, AR and ARC added together (Effigy S4), and this was greater than other combinations and than any of the state calls individually. H3K27Ac is the but marker common to all these states, suggesting that H3K27Ac is the most informative predictor of enhancers.
Since H3K4me1 does non improve predictions and is the but thing that distinguishes between AR and EAR (by its presence or absence), an improved model would consolidate AR and EAR into a single state and reassign "1" to H3K4me1 instead of "iii", leaving this mark exclusively to define weak (or primed) promoters.
To validate our method of enhancer prediction, we compared our predictions with ENCODE Encyclopedia, Version 3 (zlab-annotations.umassmed.edu), EnhancerFinder, RFECS, DELTA, CSIANN, and REPTILE33–37 for held-out data using AUPRG (Effigy S5)38.
Our predictions are comparable to the Encode model that uses H3K27Ac overlapping with distal DHS, RFECS and REPTILE, which had the lowest average rank across tissues (Table iii, Figure S5). Our predictions compared favorably to EnhancerFinder and CSIANN which had an boilerplate rank > 6 across the different tissues; centre, midbrain, hindbrain, neural tube and limb. Predictions are merely available for these tissues. Thus, StatePaintR ranking is useful for drawing quantitative comparisons between dissimilar models, making predictions, or prioritizing regions for functional evidence.
Discussion
We created a platform for hosting, browsing, and generating new genome annotation models called StateHub. The StateHub framework makes information technology possible to specify combinations of genomic information as they relate to regions of functional significance in epigenetically marked chromatin. In addition, we created a software package, StatePaintR, that facilitates the use of StateHub models to generate browser tracks for bioinformatic analyses. We showed how StatePaintR tin can be used as office of a workflow with uniformly processed data to generate reproducible annotations from public and private data sources.
Our framework does not replace electric current machine learning methods, the aim of which is to discover states. Only these methods suffer from certain drawbacks that nosotros take addressed with a dominion-based approach that provides greater transparency and reproducibility. For example, it is often the case with machine-learning methods that more states are discovered than immediately understood, and there take been different solutions proposed. During discovery, one could iteratively reduce the number of states, minimizing the number of similar or redundant combinations of histone marks. So the number of discovered states would depend on the number of unique data types used for learning and their distribution effectually known features. This procedure makes replication in different settings (in different labs or with different types of experiments) nearly impossible. Our method avoids these issues, allowing users to specify a model of the epigenome in a matrix (as in Figure 1) that accounts for all known possibilities. Thus, we congenital a comprehensive framework for a rule-based annotation, reflecting current hypotheses (or models) of the epigenome.
A significant drawback of our arroyo is that some unusual combinations of marks that may have biological office will be ignored. This has much to do with the fact that StatePaintR is not for discovering novel states, but rather for annotating the genome according to a specific, existing model. Nonetheless, the label assignment step of other chromatin state discovery tools also suffers the same limitations; states are aggregated or optimized in an iterative fashion based on prior knowledge and assumptions. ENCODE for example has published tracks for both ChromHMM and Segway that include multiple states with similar names (e.grand. "Tss" vs. "TssF" from ChromHMM, and "EnhF1" vs. "EnhF3" from Segwaytwenty). To resolve discrepancies betwixt the two methods, the authors of those studies proposed a combined analysis to simplify the number of state labels and summarize discovery using a rule-based metric not unlike a StateHub model. Thus, they classified regions into 7 types "emphasizing biologically meaningful differences"xx. In direct comparisons, we found that our ain annotations exhibited greater similarity to the combined analysis than to either of the Segway or ChromHMM tracks separately (not shown). Any the protocol, the bones problem persists; machine-learning is able to provide insight into what the categories are, but not how many categories there should be. Currently this remains the exclusive province of the biologist.
Ane of the additional challenges is compatibility between information sets. In order for two or more than cell types to exist annotated according to the same model, it is necessary to combine each of the cell types for the training stride. Ane solution is concatenation of genomesxx. Another approach is to jointly model epigenomes in parallel, as proposed in Integrative and Discriminative Epigenome Annotation System (IDEAS)39. This approach has the distinctive advantage of besides modeling segment boundaries. Our arroyo does not model boundaries, but does offering some advantages. One is reproducibility: StatePaintR ever produces the aforementioned note independently for each cell type from the same model. Secondly, even samples with different types of data or missing data result in compatible annotations considering they come from the same model. Third, the models, composed of a 2D matrix with a range of 4 values, are relatively easy to sympathise and author. Every file produced in StatePaintR contains a tape of the model ID, genome version and all the source files. Clinicians working with human genetics volition value consistency and reproducibility across datasets. We produced annotations for REMC, ENCODE, IHEC and pattern and fabricated these available on the StateHub website for the two models described in this paper. The website besides has links to browser sessions where they can exist explored and used to create figures. A fourth advantage is speed: samples tin be processed in parallel and there is no computationally expensive learning stride, allowing a typical sample to be annotated in 15 seconds (Figure S1).
A final feature that is very useful is the ranking past peak score (Figure S5). Using this scheme, we investigated what states contribute most to true enhancers (Figure S2–Figure S4). We found that H3K27Ac divers the best predictive subset of annotations for VISTA enhancers. We also investigated unlike approaches for handling multiple peak calls for a land and constitute the median to be optimal (Figure S2), and incorporated this method as the default beliefs of StatePaintR. When we compared our predictions to held-out information, they were comparable to the best enhancer predictions34,37 and ENCODE enhancers26 and on the web (unpublished).
We demonstrated a workflow wherein new models generate annotations, which are used to test predictions against experimental data, and so in plough to make improvements to old models. We anticipate that this will exist valuable in testing new ideas and hypotheses generated from unsupervised methods. The ability to rank features also aids in prioritizing variants for GWAS and studies of somatic mutations. Knowing which variants overlap features in the epigenomic landscape of a particular cell type is cardinal. In the future, other methods may become bachelor for incorporation into StatePaintR but the models described in StateHub volition remain stable.
Conclusions
Nosotros introduced 2 new computational resource, an online database of chromatin country models and processed genome segmentations called StateHub, and an R/Bioconductor tool called StatePaintR, which translates epigenomics files into segmentations using these models. One may annotate incomplete datasets rapidly and sensibly according to a single model specification that gracefully degrades to lesser annotations with missing data. Annotations accept header documentation with genome version, StateHub model, and the names of source files and their mappings. These tools certificate segmentations and state labels precisely equally they are used in individual studies and to allow comparisons betwixt evolving models of epigenomic states as they relate to NGS experiments. They too enable mixing of epigenomic states with other types of data, such as 3D looping assays, transcription factors, primary sequence features such as position weight matrices, or disease variants.
Software availability
StateHub bachelor from: http://statehub.org/
Archived source code of StateHub as at time of publication: https://zenodo.org/record/1148792forty
StatePaintR available from: http://www.bioconductor.org/packages/release/bioc/html/StatePaintR.html
Source code of StatePaintR availabe from: http://www.github.com/Simon-Coetzee/StatePaintR
Archived source lawmaking of StatePaintR as at fourth dimension of publication: https://zenodo.org/record/113782541
License: GPL v3.0
At the time of publication we accept submitted our package to Bioconductor. A new version of the article volition exist updated one time this bundle is bachelor. For now, the unabridged package is available on GitHub
Information availability
The following are additional files containing manifests to run StatePaintR with electric current releases of all public datasets listed in Table two, links to sectionalization tracks, and all lawmaking used for analysis and generation of figures in this manuscript. Complete code generated from R markdown (Rnotebooks/html format) for generating all analyses, figures and tables is available here.
Supplementary material, including Supplementary File 1 and Supplemental Figures i–5 are available on figshare here:
https://doi.org/10.6084/m9.figshare.1219508725
Supplementary File 1: Statepaintr.nb.html: This file contains code for all the examples and employ cases in the text of this manuscript, generated as an html from Rmarkdown.
Figure S1: Relationship between data and runtime. StatePaintR takes simply a few seconds to run. The exact time depends on the number number of unique segments (lines of data) created past overlapping genomic intervals of all input files, cumulative. Thus, 128 Roadmap tissues can be run in ten sec × 128 ≈ one,280 sec (21 min).
Figure S2: Predictions with multiple marks. Ranked ChIP-seq peak scores for multiple marks were used to rank active enhancers (H3K4me1 + H3K27Ac + DHS) by three methods (median, mean, ceiling) and compared to a sample (northward = 100) of experimentally validated enhancers. The boilerplate or median of three marks was a better predictor than ceiling. The choice of role is subservient to choice of information for ranking–if one of the three is less informative, it will produce false positives when using the max method–therefore it is better to eliminate uninformative marks. Meet also Figure S4.
Figure S3: Ranking enhancers with subsets of marks. Combinations of marks were used to predict agile enhancers past the max ranking method (as in Effigy S2) and compared to enhancer score. "All" includes regulatory (H3K4me1), active (H3K27Ac), and core (DHS). We also tried a leave-one-out strategy for each of these categories in succession. Leaving out H3K4me1 ("no regulatory") produced superior predictions, suggesting that its inclusion made the predictions less specific.
Effigy S4: Chromatin states as predictors of true enhancers. We tested different chromatin states for their ability to predict true enhancers under the poised focused promoter model. Active enhancers exhibited the greatest predictive power under the precision recall gain bend.
Figure S5: Performance of enhancer predictions. Area under precision-recollect-gain curves reflect the accuracy of three models of enhancer prediction. True positive enhancers are those validated in the VISTA enhancer browser. The ENCODE method (in blue) and the StatePaintR method (in blood-red) show like accuracy in retrieving VISTA enhancers showing tissue specific enhancer activeness, while EnhancerFinder (in green) is less accurate.
Grant data
Copyright
Open Peer Review
Current Reviewer Status: Central to Reviewer Statuses VIEW HIDE CanonicalThe paper is scientifically sound in its electric current form and simply minor, if any, improvements are suggested Approved with reservations A number of small changes, sometimes more meaning revisions are required to address specific details and improve the papers academic merit. Non approvedFundamental flaws in the paper seriously undermine the findings and conclusions
Version ii
VERSION 2
PUBLISHED 07 May 2020 Revised
Reviewer Report29 May 2020
Maxwell W. Libbrecht , School of Computing Sciences, Simon Fraser Academy, Burnaby, BC, Canada
Approved with Reservations
VIEWS 0
Reviewer Report28 May 2020
Yongjin Park , Broad Institute, Massachusetts Institute of Technology, Harvard Academy, Cambridge, MA, United states; Department of Pathology and Statistics, The University of British Columbia, Vancouver, BC, Canada
Approved with Reservations
VIEWS 0
Reviewer Report26 May 2020
Guillaume J. Filion , Gene Regulation, Stem Cells and Cancer Programme, Heart for Genomic Regulation, The Barcelona Found of Science and Technology, Barcelona, Spain
Approved with Reservations
VIEWS 0
Version 1
VERSION 1
PUBLISHED 22 Feb 2018
Reviewer Study23 May 2018
Guillaume J. Filion , Gene Regulation, Stalk Cells and Cancer Plan, Heart for Genomic Regulation, The Barcelona Institute of Science and Applied science, Barcelona, Spain
Approved with Reservations
VIEWS 0
Reviewer Reportx May 2018
Yongjin Park , Wide Constitute, Massachusetts Institute of Technology, Harvard Academy, Cambridge, MA, United states; Department of Pathology and Statistics, The University of British Columbia, Vancouver, BC, Canada
Approved with Reservations
VIEWS 0
Reviewer Written report30 April 2018
Maxwell W. Libbrecht , Schoolhouse of Computing Sciences, Simon Fraser University, Burnaby, BC, Canada
Approved with Reservations
VIEWS 0
Source: https://f1000research.com/articles/7-214
Comments on this article Comments (0)
Version two
VERSION 2 PUBLISHED 22 Feb 2018
Comment